生化学における遺伝子工学的技術 その2
目 的
PCR法の開発によってきわめて微量の試料から DNA を抽出し、オリゴ DNA プライマーを用いて目的のDNA 領域を短時間内にしかも容易に増幅することが可能になった。遺伝子工学技術はPCR法が開発されてから飛躍的に進歩し、医学の診断を含め広い分野で応用されている。ここでは、PCR法の原理とその医学応用について学ぶ。
7)PCR法によるDNAの増幅
8)PCR法による遺伝子変異の検出
7)PCR法によるDNAの増幅
ここでは、ホスホリパーゼD2(PLD2)遺伝子の増幅を確かめる。
準 備
- テンプレート DNA:鋳型となる2本鎖 DNA
- オリゴプライマー:目的のDNAに相補的なオリゴDNA
- PCR用緩衝液、dNTP、Taqポリメラーゼ、ミネラルオイル
- PCR用滅菌マイクロチューブ(500 μl用)
- アガロースゲル電気泳動用緩衝液:0.5x TBE 濃縮緩衝液(5x TBE)を10倍希釈し、泳動用とアガロースゲルの調製に用いる。
- 1%アガロースゲル:アガロース1g を秤量し、100 ml の0.5x TBEを三角フラスコに入れる。オートクレーブあるいは電子レンジでゲルを完全に溶かす。室温に放置するか、流水中で50-60℃(大体手で触れられるくらい)に冷やす。冷やしすぎるとゲルが固まるので、時々三角フラスコを振ってゲルが固まらないように注意する。エチジウムブロマイド(ethidium bromide)を終濃度0.5μg/ml となるように加える。型(ゲルモールド)に溶解したアガロースを流し込み、室温に放置して固める。ゲルが十分に固まったところで、ゲルを壊さないように注意しながらゆっくりとコーム(櫛形)を抜く。
- ピペットマン(P-20)、滅菌チップ および滅菌マイクロチューブ、電気泳動漕
実 験 9 PCR法によるDNAの増幅
- PCR用マイクロチューブを用意し、それぞれに以下のものを入れる。
|
| テンプレートDNA (1 μg) |
| 10倍濃縮緩衝液 |
| 上流(5'側)プライマー |
| 下流(3'側)プライマー |
| dNTP (各200 μM) |
| Taqポリメラーゼ |
|
| 1 μl |
| 5 μl |
| 1 μl |
| 1 μl |
| 1.25 μl each |
| 13 μl |
|
- ミネラルオイルをこのチューブに1滴おとし、PCR装置にチューブをセットする。
- 93℃, 1分、55℃, 2分、72℃, 2分の温度変化サイクルをそれぞれ20, 22, 24, 26, 28回繰り返すようにセットし、反応を開始する。また、反応のコンポーネント(テンプレートDNA、プライマー、dNTP、Taqポリメラーゼ)のどれか1つを含まないマイクロチューブも用意し、28回のサイクルで増幅を行う。
- ピペットマンのチップの先端をチューブの底に入れて 10 μl 取り、色素液 2 μl と混ぜてアガロースゲル電気泳動を行う。別のウェルに分子量マーカーを入れて同時に泳動する。
(今回の実習ではすでにPCRサンプルと色素液をミックスしてあるので、チューブから10 μl をP-20のピペットマンで取り、以下の図のようにそれぞれのウェルにサンプルを入れ30分間電気泳動を行う。)
- 電気泳動後、UV照射によってDNAの存在を確認し、写真を撮る。サイクルの変化によりのDNAバンドがどのように変化するか、テンプレートDNAあるいはdNTPを反応液中に入れなかったサンプルではどのような変化が起こったか確認する。

図 7-1
7.2)RT-PCR法によるRNAの解析
PCR法は DNAを試験管内で増幅する方法であり、RNAを鋳型とした増幅はできない。しかし、逆転写酵素によりRNAからcDNAを合成することにより、PCR法を RNAの解析に応用することができる。この方法により、少量の材料からRNAレベルでの遺伝子発現の解析やRNAをシークエンスすることが可能になった。
1)RNAの調製
RNase は汗や唾液などにも存在し、RNAの調製時には必ずプラスチック手袋を着用し、必要以上に話をしないことが大切である。特に気合いを入れ、集中して実験を行うようにする。器具は、滅菌済みプラスチック製品を用いる。
RNaseを速やかに変性させるグアニジン (GTC) やフェノールで手早く組織を処理するために、いろいろな方法が考案されているが、きれいなRNAをとるためには多少の熟練を要するので、実習では行わない。
2)逆転写酵素(Reverse Transcriptase, RT)反応
上記のように、RNAの取り扱いには十分注意する。cDNA合成のプライマーとしては、オリゴ(dT)、ランダムヘキサマー(dN6)、あるいは目的遺伝子の3’側プライマーが使われる。まず、RNAとプライマーを十分に結合させ(アニーリング)、逆転写酵素を加えて反応を開始する。
実 験 10 RTによるcDNAの合成
- PCR用マイクロチューブに以下のものをいれる。
|
| 組織より調製したRNA (1 μg) |
| ランダムヘキサマー(dN6) |
| 滅菌水 |
|
|
|
| A+B=10μl となるように調整し、全体で11.5 μl とする。 |
|
- 5分間37℃で反応後、速やかに氷に入れ10分間放置する。
- 以下のものを加え、42℃で60分間反応させる。
|
| 5倍濃縮緩衝液 |
| dNTPs (2.5 mM) |
| SuperScript RT |
|
|
- 95℃で5分間加熱し、RTを変性させる。
実 験 11 PCRによるcDNAの増幅
- PCR用マイクロチューブに以下のものを入れる。
|
| テンプレートcDNA |
| 10倍濃縮緩衝液 |
| 上流(5'側)プライマー |
| 下流(3'側)プライマー |
| dNTP (各200 μM) |
| Taqポリメラーゼ |
|
| 5 μl |
| 5 μl |
| 1 μl |
| 1 μl |
| 1.25 μl |
| 0.25 μl |
|
- 以下実験9と同様の操作を行う。
→Top
8)PCR法による遺伝子変異の検出
遺伝病やがんなど、疾患に関与する遺伝子異常は遺伝子の増幅、再編成、欠失などゲノムレベルでの規模の大きい異常と、点突然変異、数塩基対の欠失・挿入といった微細なレベルの異常とに大別できる。ヒトDNAの多型も、多くは塩基配列の微視的な相違に基づいている。
比較的大きな欠失・挿入では電気泳動により目的とするDNA断片の大きさを比較して、変異を同定することが可能である。しかし、点突然変異のようにDNA断片の大きさに変化がない場合には、当然のことながら大きさの比較では変異は検出できない。特定のDNA断片内の微視的な変異を同定するためには、その全塩基配列を決定すればよい。しかし、PCRを応用したいくつかの塩基配列決定法が開発されているが、時間と労力を必要とし、多数のサンプルの広範な解析には実用的ではない。特に目的の遺伝子が大きな場合には全塩基配列の決定にはかなりの時間を要する。
多数のサンプルの広範な解析を目的として、いくつかの方法が開発されている。ここでは、制限酵素の認識配列に変異が起こった場合に応用できるPCR-RFLP(Restriction Fragment Length Polymorphism)法を行い、さらに1本鎖DNA(ssDNA)の高次構造の違いにより変異を同定するPCR-SSCP(Single Nucleotide Conformation Polymorphism)についてその原理と方法を理解する。
8.1)PCR-RFLP法
制限酵素の塩基配列の認識(表7−1参照)は厳密であり、認識配列の1塩基でも異なるともはや酵素は認識しなくなる。遺伝子上の制限酵素認識配列に変異が起きた場合、その変異部位を含むDNA断片をPCR法により増幅し、その後制限酵素処理をして電気泳動により分離すると、元のDNA断片(制限酵素により分解)と変異DNA(切断されない)は大きさが異なり、遺伝子異常の有無を簡便に検出できる。逆に、制限酵素により認識されなかった配列に変異が生じて新たな制限酵素認識配列が出現した場合も、この方法により解析可能である。
ここでは、MAPK遺伝子上のXbaI認識配列上に認められた点突然変異をPCR-RFLP法により解析する。
準 備
- テンプレート DNA:鋳型となるds DNA
- オリゴプライマー:目的のDNAに相補的なオリゴDNA
- PCR用緩衝液、dNTP、Taqポリメラーゼ、ミネラルオイル
- PCR用滅菌マイクロチューブ(500 μl用)
- 制限酵素(XbaI)、制限酵素用10倍濃縮緩衝液 (X10 H) および滅菌超純水
- 色素液:30% グリセロール、0.25% ブロモフェノールブルー(BPB)、0.25% キシレンシアノール(XC)
- DNA マーカー:λ DNA/Hind III , ΦX174 DNA/Hae III
- アガロースゲル電気泳動用緩衝液:0.5x TBE
- 1%アガロースゲル
- ピペットマン(P-20)、滅菌チップ および滅菌マイクロチューブ、電気泳動漕、37℃インキュベーター
実 験 12 PCR法によるDNAの増幅
- PCR用マイクロチューブを用意し、それぞれに以下のものを入れる。
|
| テンプレートDNA (1 μg) |
| 10倍濃縮緩衝液 |
| 上流(5'側)プライマー |
| 下流(3'側)プライマー |
| dNTP (各200 μM) |
| Taqポリメラーゼ |
|
| 1 μl |
| 5 μl |
| 1 μl |
| 1 μl |
| 1.25 μl |
| 0.25 μl |
|
- ミネラルオイルをこのチューブに1滴おとし、PCR装置にチューブをセットする。
- 93℃, 1分、55℃, 2分、72℃, 2分の温度変化サイクルを28回繰り返すようにセットし、反応を開始する。
実 験 13 制限酵素処理
- 滅菌マイクロチューブに以下のものを入れる。
|
| PCR反応液 |
| 10倍濃縮緩衝液 (X10 H) |
| 制限酵素 (XbaI) |
| 減菌超純水 |
|
|
- マイクロ遠心機で 12,000 rpm, 数秒間遠心する。
- 37℃, 1時間インキュベートする。
- 色素液 4 μlをマイクロチューブに加え、 12,000 rpmで数秒間遠心する。
- 試料10 μlを[1%アガローズゲル上で電気泳動する。
- 電気泳動後、UV照射によってDNAの存在を確認し、写真を撮る。サイクルの変化によりのDNAバンドがどのように変化するか、テンプレートDNAあるいはdNTPを反応液中に入れなかったサンプルではどのような変化が起こったか確認する。

図 7-2

図 7-3
8.2)PCR-SSCP法
PCR反応を用いて目的とする塩基配列を増幅し、そのDNA断片を1本鎖(ssDNA)に変性したときDNA断片内の塩基配列が1塩基でも異なっている時は別の高次構造をとり電気泳動上での移動度が異なる。塩基配列を解読することに比べれば精度は落ちるが、簡便で短時間に大量のサンプルの解析が可能であることから、スクリーニングに用いられる。
実際には、まずDNA断片目的とするをPCR法により増幅する。得られた二本鎖DNA断片(dsDNA)を加熱により変性させ一本鎖(ssDNA)とした後に、非変性ポリアクリルアミドゲル電気泳動にて分離する。変性して1本鎖となったDNA断片は、鎖内(分子内)水素結合や塩基間の相互作用などによりその塩基配列に特異的な高次構造をとる。1塩基でも変異があると、その1本鎖DNA断片の高次構造は元の1本鎖DNA断片の高次構造とは異なり、電気泳動上は異なる位置に泳動される。
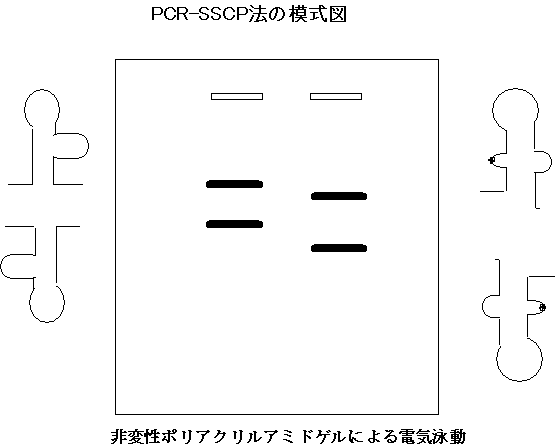
図 7-4
→Top